3D Cell visualization
In this tutorial, we will make 3D visualization of the graph components that constitute cells in MPX data using R.
Setup
library(dplyr)
library(tidyr)
library(here)
library(ggplot2)
library(pixelatorR)
library(stringr)
library(SeuratObject)
Load data
We will continue with the stimulated T cell data that we analyzed in the previous tutorial (Karlsson et al., Nature Methods 2024).
DATA_DIR <- "path/to/local/folder"
Sys.setenv("DATA_DIR" = DATA_DIR)
pg_data_combined <- readRDS(file.path(DATA_DIR, "uropod_spatial.rds"))
pg_data_combined
An object of class Seurat
3400 features across 786 samples within 4 assays
Active assay: mpxCells (80 features, 80 variable features)
1 layer present: counts
3 other assays present: polarization, colocalization, mpx
2 dimensional reductions calculated: pca, umap
Now we can select components with high colocalization for a specific marker pair to get some good example components to work with. First, we will filter our colocalization scores to keep only CD162/CD50 and then we will add the abundance data for CD162, CD37 and CD50 to the colocalization table.
# Make sure that we are using the correct assay
DefaultAssay(pg_data_combined) <- "mpxCells"
colocalization_scores <-
ColocalizationScores(pg_data_combined, meta_data_columns = "sample") %>%
unite(contrast, marker_1, marker_2, sep = "/") %>%
filter(contrast == "CD162/CD50") %>%
left_join(y = FetchData(pg_data_combined, vars = c("CD162", "CD50", "CD37"), layer = "counts") %>%
as_tibble(rownames = "component"),
by = "component")
Next, we will select components with high colocalization scores for CD162/CD50. In the histogram below, we see the distribution of Pearson Z scores for the CD162/CD50 colocalization. Since we want to narrow down the selection of components, we set a threshold for the Pearson Z scores. Here, we use a threshold of 35 to keep the components with the highest colocalization scores. Note that the threshold is arbitrary and should be adjusted based on the data.
# Set Pearson Z threshold
pearson_z_threshold <- 35
ggplot(colocalization_scores, aes(pearson_z)) +
geom_histogram(bins = 50) +
geom_vline(xintercept = pearson_z_threshold, linetype = "dashed") +
labs(title = "Pearson Z scores [CD162/CD50]")
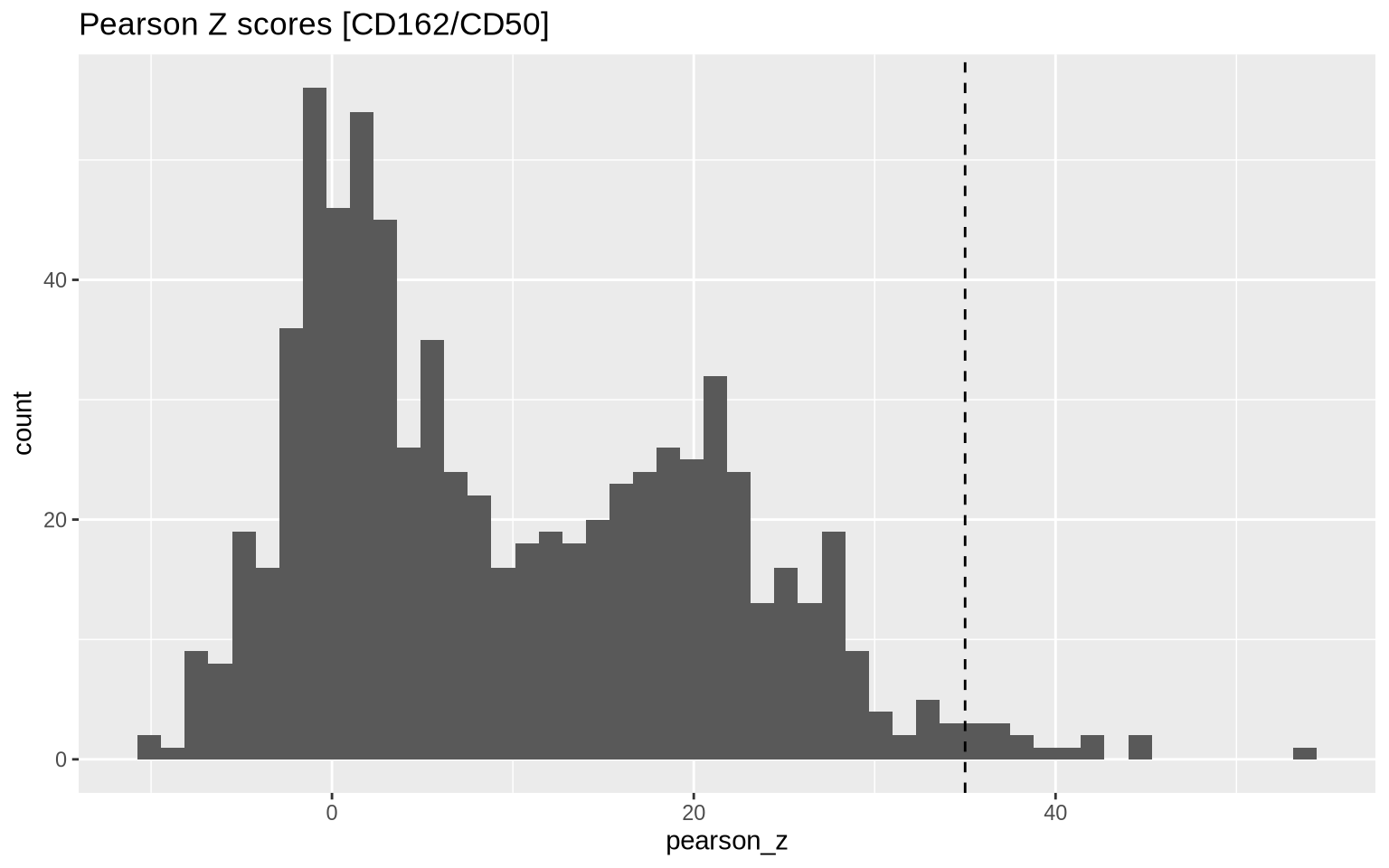
Then we apply our threshold to get a final selection of 13 components.
plot_components <-
colocalization_scores %>%
filter(pearson_z > pearson_z_threshold, sample == "Stimulated") %>%
arrange(-CD162) %>%
select(component, pearson_z, CD162, CD50, CD37)
plot_components
# A tibble: 15 × 5
component pearson_z CD162 CD50 CD37
<chr> <dbl> <dbl> <dbl> <dbl>
1 Stimulated_61cb84c709f41e7b 41.7 947 1222 631
2 Stimulated_59b284adcc91ec91 44.1 789 864 448
3 Stimulated_23606c9599265d9a 40.1 690 707 452
4 Stimulated_18278b69dc73ad16 41.7 486 736 249
5 Stimulated_437d692c40d6d601 53.8 450 493 286
6 Stimulated_50cd2ae85ea1c0e1 44.6 414 651 287
7 Stimulated_19ab7abc50018ac8 40.5 303 467 116
8 Stimulated_898664166bbd71ae 35.6 301 626 317
9 Stimulated_d33f7aa55f7874ff 36.5 220 238 112
10 Stimulated_7778298975953f4f 36.9 216 420 138
11 Stimulated_703a192bee0b36f2 38.0 197 183 178
12 Stimulated_f4d9b2d31012177a 38.4 178 161 119
13 Stimulated_bd77e6c5f6e229e5 36.7 153 289 108
14 Stimulated_60523a26c6f19593 35.9 136 111 38
15 Stimulated_8a91a136efcae912 35.4 68 118 69
Visualize single cell graph components in 3D
As one of the main features of the MPX technology, we can create
three-dimensional layouts to visualize single cells. The layout
algorithms wpmds and fr that we used in 2D Cell
Visualization can also be used
here.
Pre-computed layouts
As we learned in the 2D visualization tutorial, we can use pre-computed
layouts if they are available in our PXL files. Alternatively, we use
the ComputeLayout method to generate 3D layouts after we have loaded
our component graphs with LoadCellGraphs. For the latter option, we
need to set dim = 3 to make sure that the layout is computed in 3D:
# Run this to load graphs and compute 3D layouts
pg_data_combined <- pg_data_combined %>%
LoadCellGraphs(cells = plot_components %>%
pull(component)) %>%
ComputeLayout(dim = 3, layout_name = "wpmds")
In this tutorial, we load the pre-computed layouts from the PXL files.
pg_data_combined <- pg_data_combined %>%
LoadCellGraphs(cells = plot_components %>%
pull(component),
force = TRUE,
load_layouts = TRUE)
Now when we have the 3D layouts, we can visualize component graphs as 3D
scatter plots where each point corresponds to a UPI. Just as in the 2D
plots, we can color the points based on marker abundance to see how it
is distributed on the cell surface. However, now that we have a 3D
layout, we get an interactive plot that we can rotate, pan and zoom.
Since the 3D scatter plots are quite complex, Plot3DGraph only
displays the layout for a single component.
From our table above, we can select any component. The “Stimulated_RCVCMP0001141” cell is a good candidate to visualize in 3D as it has a high Pearson Z score for CD162/CD50 and high abundance of CD162, CD50 and CD37. If we project the abundance of one of the uropod markers (CD162) on the nodes, we see that counts are indeed clustered to a small area of the cell surface.
Plot3DGraph(pg_data_combined,
cell_id = "Stimulated_898664166bbd71ae",
marker = "CD162",
layout_method = "wpmds", node_size = 3,
colors = colorRampPalette(colors = c("gray80", "orangered"))(20)) %>%
plotly::layout(scene = list(xaxis = list(title = "", showgrid = F, nticks = 0, showticklabels = F),
yaxis = list(title = "", showgrid = F, nticks = 0, showticklabels = F),
zaxis = list(title = "", showgrid = F, nticks = 0, showticklabels = F)))
We can also normalize the 3D coordinates to a unit sphere.
Plot3DGraph(pg_data_combined,
cell_id = "Stimulated_RCVCMP0001141",
marker = "CD162",
layout_method = "wpmds", node_size = 3,
project = TRUE,
colors = colorRampPalette(colors = c("gray80", "orangered"))(20)) %>%
plotly::layout(scene = list(xaxis = list(title = "", showgrid = F, nticks = 0, showticklabels = F),
yaxis = list(title = "", showgrid = F, nticks = 0, showticklabels = F),
zaxis = list(title = "", showgrid = F, nticks = 0, showticklabels = F)))
Viewing colocalization of markers
We can also visualize the colocalization of two markers in 3D. Here, we visualize CD162 and CD50 on the same MPX cell component. We use the same approach as above, but now we create two plots and combine them in one view.
# Create a plot for CD162
plotly_obj_cd162 <- Plot3DGraph(pg_data_combined,
cell_id = "Stimulated_898664166bbd71ae",
marker = "CD162",
layout_method = "wpmds", node_size = 3,
colors = colorRampPalette(colors = c("gray80", "orangered"))(20),
scene = "scene") %>%
plotly::colorbar(x = -0.1, y = 1) %>%
plotly::layout(title = list(title = ''))
plotly_obj_cd162$x$layout$annotations <- NULL
# Create a plot for CD50
plotly_obj_cd50 <- Plot3DGraph(pg_data_combined,
cell_id = "Stimulated_898664166bbd71ae",
marker = "CD50",
layout_method = "wpmds", node_size = 3,
colors = colorRampPalette(colors = c("gray80", "orangered"))(20),
scene = "scene2") %>%
plotly::colorbar(y = 1)
plotly_obj_cd50$x$layout$annotations <- NULL
# Combine plots
main_plot <- plotly::subplot(plotly_obj_cd162,
plotly_obj_cd50, nrows = 1, margin = 0.06) %>%
plotly::layout(scene = list(domain = list(x = c(0, 0.5),
aspectmode = "cube")),
scene2 = list(domain = list(x = c(0.5, 1),
aspectmode = "cube"))
) %>%
plotly::hide_legend()
main_plot$x$layout[1:2] <- NULL
# Add marker annotations
annotations = list(
list(
x = 0.2,
y = 1.0,
text = "CD162",
xref = "paper",
yref = "paper",
xanchor = "center",
yanchor = "bottom",
showarrow = FALSE
),
list(
x = 0.8,
y = 1,
text = "CD50",
xref = "paper",
yref = "paper",
xanchor = "center",
yanchor = "bottom",
showarrow = FALSE
)
)
main_plot <- main_plot %>% plotly::layout(annotations = annotations)
# We can sync the plots with some custom JavaScript code
main_plot %>%
htmlwidgets::onRender(
"function(x, el) {
x.on('plotly_relayout', function(d) {
const camera = Object.keys(d).filter((key) => /\\.camera$/.test(key));
if (camera.length) {
const scenes = Object.keys(x.layout).filter((key) => /^scene\\d*/.test(key));
const new_layout = {};
scenes.forEach(key => {
new_layout[key] = {...x.layout[key], camera: {...d[camera]}};
});
Plotly.relayout(x, new_layout);
}
});
}")
<IPython.lib.display.IFrame object at 0x7f9cac832f90>